The Gage Lab is focused on linking crop genomics and phenomics to develop more resilient, productive crop varieties.
Current projects:

Genotype-by-environment (GxE) interactions are a significant source of phenotypic variation. These interactions influence not only whole-plant traits but also molecular phenotypes such as gene expression. Advancing crop improvement and enhancing resilience to climate change requires a deeper understanding of how genotype and environment jointly shape transcriptomic responses. Through multi-environment RNA-seq datasets, we can investigate expression GxE, uncover its underlying mechanisms, and determine its consequences for phenotypic plasticity. This project, headed by PhD candidate Ty Thomas, aims to generate insights that ultimately support more precise, climate-resilient plant breeding and genetic strategies.
Some of the questions we are exploring:
- What are the primary sources of variation in transcript abundance, and what is the relative contribution of GxE to transcript-level variation?
- How does expression GxE influence whole-plant traits such as yield, flowering time, and plant height?
- What proportion of eQTLs are sensitive to GxE and do these effects underlie GxE for important performance phenotypes?
- Does the impact of GxE differ across different levels of the central dogma (DNA → RNA → protein → phenotype)?

Studying GxE in complex traits using quantitative genetic approaches remains experimentally and statistically challenging. Allelic differences in DNA sequences within populations can alter gene expression. Quantifying allele-specific expression (ASE) by measuring the relative mRNA abundance of each allele at heterozygous loci controls for trans-acting genetic and environmental factors, providing a robust method to characterize gene expression GxE. We quantified ASE using longitudinal RNA-seq data from diverse maize hybrids to study variation in temporal patterns of transcription. The expression patterns of these genes demonstrate gene-time interactions (GxT), which are conceptually similar to GxE. The framework for identifying genes with allelic variation for GxT will be extensible to studying GxE as well. Our aim is to use allele-specific expression analysis as a tool to identify GxE in gene expression patterns and to answer how gene expression GxE contributes to organism-level phenotypic GxE.
Some of the questions we are exploring:
- How can ASE be used to quantify gene expression GxT in maize genotypes, and how can the same approach extend to quantify gene expression GxE?
- How does allelic variation in cis-regulatory sequences contribute to gene expression GxE?
- Can cis-regulatory variation predict organism-level phenotypic GxE across environments?

Gene expression can be modulated at multiple levels of the central dogma, yet our understanding of these layers remains uneven. While decades of research have illuminated the molecular mechanisms that govern gene regulation at the DNA and RNA stages, far less is known about how translation is controlled on a gene-specific basis—even though this final step ultimately determines protein synthesis. Although transcriptional regulation is undeniably important, a growing body of evidence shows that translational control plays an equally critical and sometimes independent role in shaping cellular phenotypes. However, current technologies for quantifying translational changes, whether genome-wide or at single-gene resolution, remain technically demanding, expensive, and inaccessible for many research questions. These limitations have slowed progress toward fully understanding how translational regulation contributes to biological complexity, highlighting the need for more tractable, high-resolution approaches to study this essential layer of gene expression control.
To address this, we are working in collaboration with the Alonso-Stepanova lab at NCSU on developing computational tools for their new sequencing method that will expand the toolbox for studying gene-specific and genome-wide changes in translation efficiency.

Maize and sorghum are staple crops grown worldwide, yet we still know surprisingly little about how their root-associated microbiomes shape plant resilience. The GERMs research platform, led by Dr. Nate Korth, investigates how plant genetics, environmental stress, and rhizosphere microbial activity interact. By integrating plant and microbial transcriptomes, we attempt to uncover the molecular signals that coordinate plant responses to stress with changes in microbial function. This systems-level approach helps us identify host and microbial functions that influence plant resilience, and breeding targets for crops that can regulate their microbiomes for improved resilience.
Some of the questions we are exploring:
- How do plant genotype and environmental factors shape microbial community composition and function below ground? (ex. Heat stress, nitrogen deprivation)
- Do ancestors and relatives of maize, including heirloom varieties, teosinte, and sorghum, maintain strategies for microbiome recruitment and regulation lost in modern maize during domestication?
- Can we identify coordinated functions in plants and their microbiomes that play a role in plant stress adaptation?

Nearly all maize grown in the United States today are hybrid cultivars, but this has not always been the case. Prior to the first double-cross hybrid release in 1921, maize varieties were maintained through open pollination and locally adapted through selection for individual plant phenotypes. These open-pollinated (heirloom) populations harbor extensive phenotypic and genotypic diversity, yet remain poorly described and largely absent from modern breeding. Heirloom maize has increasing value in specialty markets, among chefs, organic farmers, and small-scale growers, and it may also contain novel alleles that illuminate the adaptive history of maize as it spread across the United States. Although heirloom maize from Mexico, South America, and Europe has been well cataloged, no equivalent characterization exists for U.S. heirlooms.
990 open-pollinated heirloom varieties with potentially diverse genetic diversity are being evaluated for:
- Two-year, two-location phenotypic evaluations with drone imaging, and high-throughput ear, kernel, and cob imaging
- ~2x coverage whole genome sequencing, 10 individuals per population
- RNA sequencing, 884 populations over 3 environments
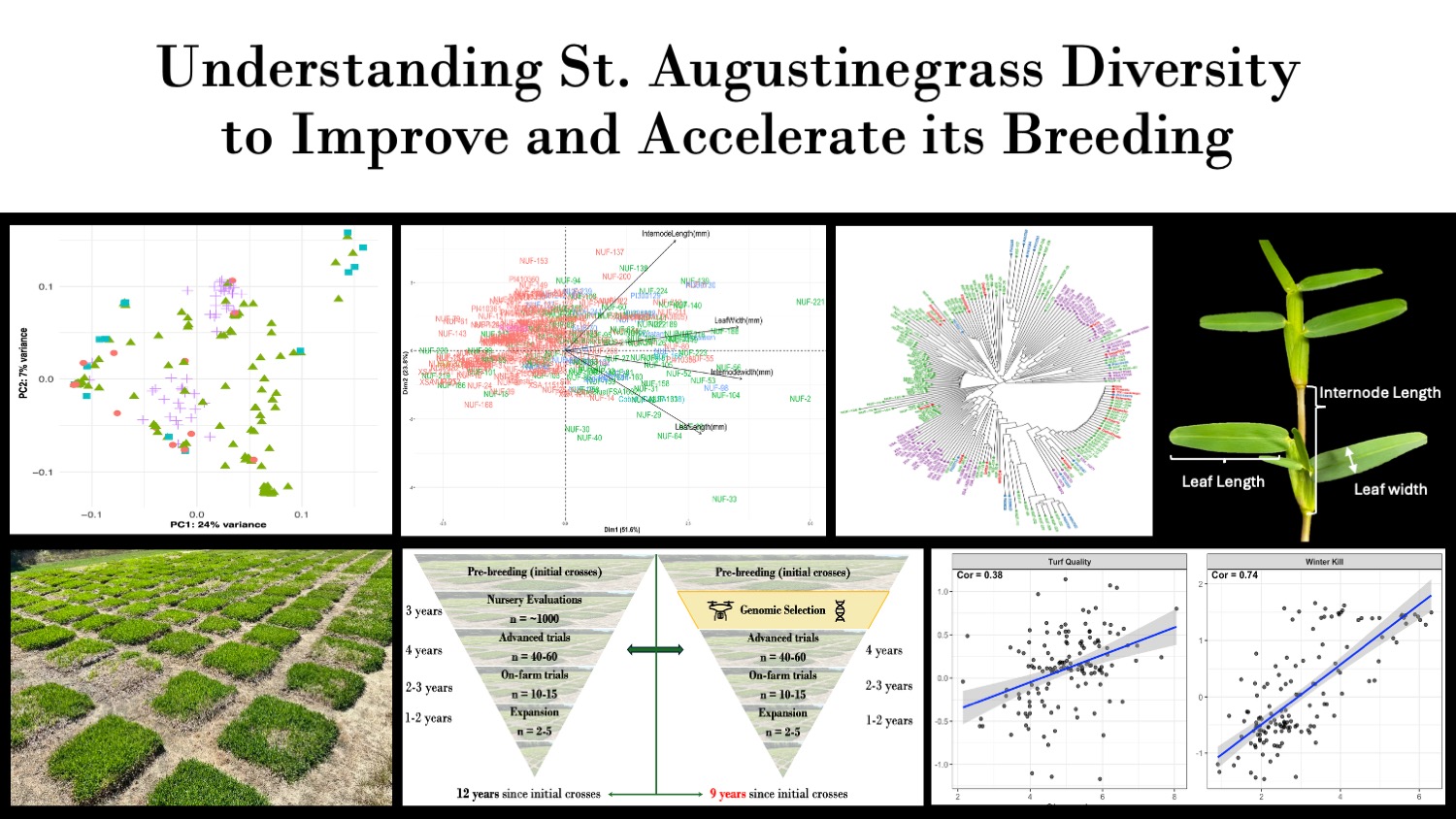
St. Augustinegrass is a warm-season turfgrass commonly found in home lawns. While we know a bit about this grass, there’s still a lot to learn about its genetics. Our research looks at the genetic diversity and population structure of St. Augustinegrass germplasm. By connecting this diversity to important traits like drought and cold tolerance, we can use genomic prediction to help breeders pick the best plants faster and more efficiently.
Some of the questions we are exploring:
- How much genetic diversity exists in St. Augustinegrass? What is the population structure among the germplasm?
- Can we identify better genotypes from germplasm to develop more resilient cultivars?
- What are the genetic factors behind important traits like drought and cold tolerance?
- Which SNPs across the genome influence these traits, and how big are their effects?
- How can genomic prediction speed up the breeding process?